Immunogenicity 101: Antidrug Antibodies, Neutralizing Antibodies, and everything in between
BY
Amber Bennett, Leo Maritz, Elizabeth Ross, Anja Schwär, Jordyn van Teylingen
CATEGORIES
TAGS
BY
Amber Bennett, Leo Maritz, Elizabeth Ross, Anja Schwär, Jordyn van Teylingen
Background
Protein-based biotherapeutic/biologic agents, such as fusion proteins and monoclonal antibodies (mAb) have become the treatment of choice for many indications (autoimmune conditions, cancers, viral infections, etc.) due to their superior efficacy and safety compared to alternative conventional therapies. However, protein therapies which require repeated administration, such as those associated with chronic conditions, are prone to triggering immune responses (Boehncke & Brembilla, 2018).
The potential for biotherapeutic agents to elicit an immune response is termed “immunogenicity” and is a common phenomenon in immunocompetent patients, despite not often being observed in pre-clinical studies, and not always having clinically relevant consequences (Kessler et al., 2006; Shankar et al., 2008). Immunogenicity can be desired – as is the case with the vaccine response (Leroux-Roels et al., 2011; Siegriest, 2018) – or it can be an unwanted reaction that reduces therapeutic efficacy, and causes adverse side effects ranging from flu-like symptoms to myelosuppression, neuropsychiatric episodes, and organ failure (Tovey & Lallemand, 2011).
Immunogenicity results in the production of anti-drug antibodies (ADAs), which bind to a biotherapeutic and sometimes completely inhibit its mechanism of action (De Groot & Scott, 2007). While it is difficult to predict immunogenic responses, certain attributes of the drug and patient characteristics can be managed to reduce the risk of ADA production.
Compared to small molecule drugs, biotherapeutics are complex entities, that are usually expressed in cellular systems, and may be unfamiliar to the human immune system. Downstream processing can cause further deviation from the expected structure or introduce contaminants, making the molecule “foreign” to the human body (Boehncke & Brembilla, 2018). Efforts to humanise biologics have been successful at reducing immunogenic responses (Pratt; 2018); however, the risk still remains, particularly with therapeutic mAbs.
In some cases, human-sequenced therapeutic mAbs contain unique, non-human sequences in the cluster differentiation region associated with immunogenicity (Harding, et al., 2010). Modifying certain amino acids in these regions could make them safer (more “human-like”), provided the efficacy of the treatment can be preserved. Intrinsic factors of the mAbs can also increase their immunogenicity, such as localisation, presence of carbohydrate sidechains, or post-translational modifications (PTMs), uncharacteristic of the natural equivalent (Kuriakose et al., 2016).
Furthermore, immunogenicity is influenced by the route of administration of a therapeutic molecule. The immune system is highly active in the subcutaneous layer of the skin, where antigen-presenting cells are concentrated. The risk of immune response is highest with subcutaneous injection, followed by intramuscular, intranasal, and intravenous routes (Kuriakose et al., 2016; Boehncke & Brembilla, 2018).
Significant effort has gone into characterisation of the ADA response at both the cellular and antibody level for a huge number of biotherapeutic agents and their biosimilars. Protein engineering and clinical protocols to increase immune tolerance of biologics are complementary approaches that need to be employed to improve treatment outcomes and minimise side effects.
Anti-Drug Antibodies
ADAs are typically classified as either neutralising or non-neutralizing (also known as binding ADAs) (Figure 1). In both cases, ADAs have a substantial influence on the pharmacokinetics and pharmacodynamics of a biotherapeutic agent. In the same way that neutralising antibodies (nAbs) neutralise pathogen infectivity (see Section 4), they can also recognize and bind to epitopes on a biotherapeutic agent, directly reducing drug availability to target molecules (Figure 1 [A] and [B]), resulting in treatment failure (El Amraniet al., 2019).
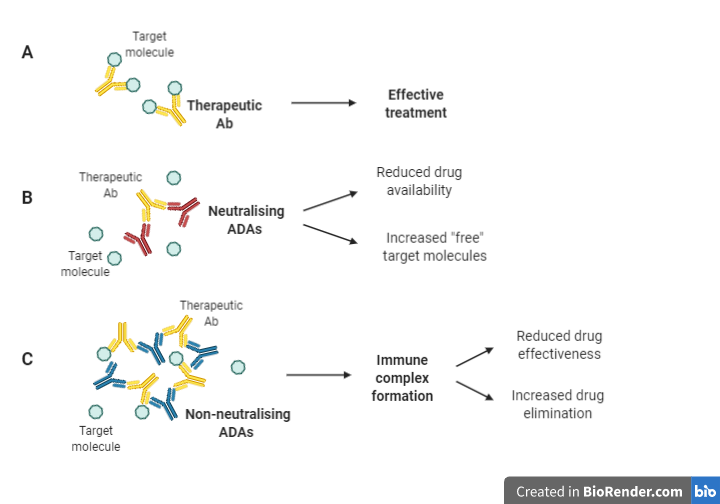
Figure 1: Mechanisms through which ADAs impact the efficacy of biotherapies (Adapted from Moussa et al. (2016); Boehncke & Brembilla (2018); El Amraniet al. (2019)).
Conversely, non-neutralizing antibodies bind to epitopes on the drug which may not directly interfere with the binding to its target but may indirectly affect drug efficacy by creating immune complexes, which decreases drug efficacy and accelerates drug clearance from circulation (Boehncke & Brembilla, 2018) (Figure 1 [C]). Furthermore, these immune complexes may be responsible for serious adverse effects (SAEs) associated with complement activation/deposition leading to cytokine release.
Biotherapeutic agents that mimic endogenous proteins may trigger collateral immune responses by eliciting cross-reaction towards similar proteins in the body. A classic example of the impact of ADAs was observed in 1998 with a recombinant Epoetin biotherapeutic agent, which was analogous to the endogenous hormone, erythropoietin (EPO). The manufactured biotherapeutic agent (of which the formula and route of administration was changed post-marketing) stimulated production of ADAs, which bound to and supressed endogenous EPO, causing some patients to experience pure red blood cell aplasia. Similarly, children receiving purified human Growth Hormone (hGH) developed ADA aggregates with the biotherapeutic as well as with endogenous hGH, resulting in impaired growth. This was rectified using recombinant hGH with reduced aggregation potential (Moussa et al., 2016).
Consequently, the detection of ADAs and nAbs in patients receiving a particular drug is important for predicting a loss in therapeutic response, and ultimately patient safety.
Detecting and Characterising an ADA response
A patient’s immunogenic response towards a given biotherapeutic agent is evaluated by developing, optimizing, and validating customized methods (most commonly immunoassays) tailored to detect and characterise ADA specific to the relevant protein drug. Validation refers to the process of demonstrating fit-for-use characteristics and optimal performance of an analytical method, in alignment with the associated regulatory bodies (e.g. U.S Pharmacopeia, U.S Food and Drug Administration, European Medicines Agency, etc.).
The first step in ADA detection involves the use of ADA screening assays, which classify samples as either ADA non-reactive (i.e. negative) or reactive (i.e. potentially positive) against an empirically determined ‘screening cut point’. Specificity confirmation assays (also known as confirmatory assays) are then performed on samples which tested ADA reactive to determine whether the ADA reactive samples detected are, in fact, true positives or non-specific reactive samples (i.e. false positives). These confirmatory assays involve establishing a ‘specificity cut point/threshold’ based on the magnitude of signal inhibition observed in samples spiked with the drug. Samples that test ADA positive may undergo further characterisation to determine antibody class/isotypes, the relative antibody levels (i.e. antibody titre) and ADA neutralising capacity.
Traditionally, nAb levels are measured with cell-based assays, which quantify the biological activity of a living cell exposed to sample material (Chatterjee et al., 2018). Cell-based formats are strongly recommended by regulatory authorities as they can more accurately show the mechanism by which nAbs exert their effects in vitro (FDA Guidelines, 2015; EMA Guidelines, 2017). These assays should be designed in a way that allow assessment of overall drug activity. In doing so, if the activity of the drug decreases when patient antibodies are added, then the antibodies can be said to have a neutralizing effect on the drug. Despite their benefits, cell-based assays can be laborious to standardise and perform, and may lack specificity due to cross-reactivity (e.g. with cytokines or other unknown factors present in patient matrices) leading to false positive results (Gupta et al., 2007).
Ligand-binding nAb assays can be used as an alternative to cell-based assays. These are easier to optimise and are more reproducible but lack the ability to elucidate the mechanism by which the nAbs exert their effect within a living biological system. In addition, to perform a ligand-binding nAb assay, the biological activity of the biotherapeutic drug product must be limited to ligand-receptor binding or protein-protein binding. In cases where the therapeutic is an enzyme, an enzymatic activity nAb assay can also be performed.
Harnessing Immunogenicity for Therapeutic Intervention
Despite the negative impact of ADAs and nAbs on the effectiveness of biologics, the immunogenic principle can be harnessed to create promising therapies for the prevention and/or treatment of disease. Therapeutic nAbs are defined by their ability to inhibit viral cellular entry or egress. In addition, nAbs can interact with and recruit other immune components, such complement, phagocytes and natural killer cells (Yasui et al., 2014). Multiple factors determine whether an antibody neutralizes a virus and provides host protection or causes Antibody Dependent Enhancement (ADE) and systemic inflammation. These include the concentration, affinity, specificity, and isotype of the nAb (Wang et al., 2014).
nAb therapy can be broadly classified as passive and active immunisation. Vaccines provide the foundation for active immunity by enabling the immune system to produce nAbs in response to an active pathogenic infection (Salazar, 2017; Siegriest, 2018). As a result, upon re-exposure to the same pathogen, the nAb response is more rapid and effective. Measuring antibody neutralising potential is critical in the development of nAb-based active immunity.
Passive immunisation, or antibody therapy, is fast becoming a rapidly acting alternative treatment to vaccines, especially for newly emerging viruses. During the outbreaks of SARS-CoV, MERS-CoV and SARS-CoV-2, convalescent plasma from patients who possessed nAbs was an effective treatment in reducing morbidity and mortality in severe cases (Ko et al., 2018). However, access to convalescent plasma is sometimes limited, and donor plasma containing non-neutralizing antibodies may cause undesirable side effects (Iwasaki & Yang, 2020). Thus, isolation and administration of nAbs represents a safer and more effective therapeutic strategy.
nAbs can render invading particles innocuous by binding to their receptor-binding domain (RBD) and/or other domains, preventing viral interaction with the entry receptors on the target cell and blocking cell entry (Figure 2 [A]). This can be done through various mechanisms, usually depending on the type of pathogen and how it enters the host cell (Forthal, 2015). Post neutralisation, the pathogen-antibody complex is taken up and degraded by macrophages. In addition, nAbs can prevent pathogens from binding to their entry receptor through competitive or non-competitive receptor binding (Figure 2 [B]). Low quality nAbs, however, bind to viral particles and to Fc receptors (FcRs) expressed on immune cells, facilitating viral entry and infection (Figure 2 [C]). Immune complexes and viral RNA can activate cytokine storms and immunopathology.
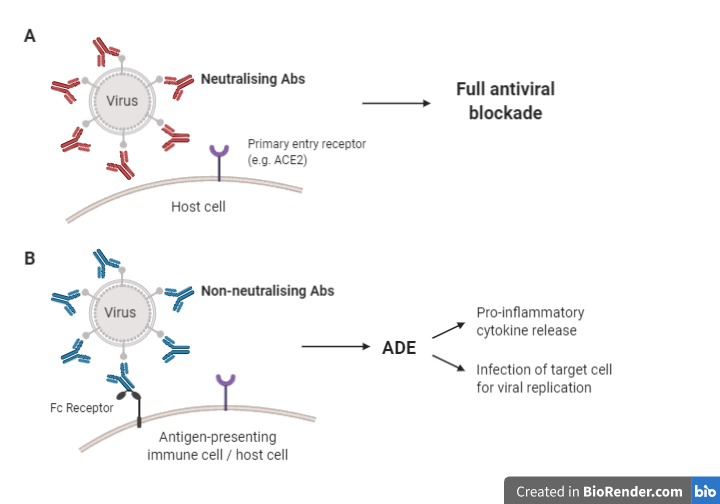
Figure 2: Potential mechanisms of action of nAbs (Adapted from Iwasaki & Yang (2020); Cao et al. (2020); (Flint et al., (2020)).
Relevant to the current SARS-CoV-2 pandemic, a Phase 1 clinical trial is recruiting hospitalized patients with moderate COVID-19 to investigate the safety and efficacy of a mAb designed to neutralise the S1 subunit of SARS-CoV-2 Spike protein. The nAb Fc region is engineered to inhibit interactions with host Fc receptors, thereby decreasing risk of ADE of SARS-CoV-2 infection (Cao et al., 2020).
Another promising nAb therapeutic, ibalizumab, was recently approved for the treatment of highly-resistant HIV-1 infection (Emu et al., 2018). While ibalizumab does not directly interact with the circulating virus as previously described, it instead targets the extracellular CD4 domain and interferes with the binding of HIV-1 to its primary receptor on target cells.
nAbs can be engineered with molecular precision, to reduce the risk of ADE development and to increase Ab affinity and specificity, thereby negating unwanted side effects (Kaplon & Reichert, 2018). In addition, nAbs can be produced on a mass-scale for widespread delivery, an important aspect to consider in the face of new novel viruses and fast-spreading pandemics.
Concluding Remarks
Long-term maintenance therapies using biotherapeutic drugs are becoming increasingly popular for many conditions, due to their high efficacy and safety. However, the administration of biotherapeutic drug products is always accompanied by a risk for an immunogenic response.
To assess the immunogenic potential of a biotherapeutic drug, and to correlate laboratory results with clinical events, it is vital to develop validated laboratory test methods that provide accurate assessment and characterisation of ADA responses. Significant efforts in characterisation of the ADA response at both the cellular and antibody level for biotherapeutic agents and their biosimilars, have allowed for increased patient safety and tolerability. Ultimately, protein engineering and clinical protocols to increase immune tolerance of biologics are complementary approaches that will need to be employed to improve treatment outcomes and minimise side effects.
Advances in antibody isolation and production technologies have enabled the development of highly potent and specific clinical Ab products for the treatment of inherent and externally mediated diseases. At present, there are more than a thousand nAb-related therapeutic candidates in the preclinical and clinical trial stages (https://clinicaltrials.gov/ct2/results?cond=&term=neutralizing+antibody&cntry=&state=&city=&dist=), highlighting the potential of antibody therapy to combat a wide range of conditions.
References:
Boehncke & Brembilla (2018): https://doi.org/10.1080/1744666X.2018.1468753
Cao et al. (2020): https://doi.org/10.1101/2020.09.27.316174
Chatterjee et al. (2018): https://doi.org/10.1016/j.jim.2017.09.004
De Groot & Scott (2007): https://doi.org/10.1016/j.it.2007.07.011
El Amrani et al. (2019): https://doi.org/10.1016/j.jtauto.2019.100004
EMA. Guideline on Immunogenicity assessment of therapeutic proteins (2017): www.ema.europa.eu/docs/enGB/documentlibrary/Scientificguideline/2017/06/WC500228861.pdf
Emu et al. (2018): https://doi.org/10.1056/nejmoa1711460
FDA Guidance on Scientific Considerations For Demonstrating Biosimilarity To a Reference Product (2015): www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291128.pdf
Flint et al. (2020): Principles of virology, 4th Edition, Volume 2. John Wiley & Sons.
Forthal (2015): https://doi.org/10.1128/9781555817411.ch2
Gupta et al. (2007): https://doi.org/10.1016/j.jim.2006.12.004
Harding et al. (2010): https://doi.org/10.4161/mabs.2.3.11641
Iwasaki & Yang (2020): https://doi.org/10.1038/s41577-020-0321-6
Kaplon & Reichert (2018): https://doi.org/10.1080/19420862.2018.1415671
Kessler et al. (2006): https://doi.org/10.1093/ndt/gfl476
Ko et al. (2018): https://doi.org/10.3851/imp3243
Kuriakose et al. (2016): https://doi.org/10.1155/2016/1298473
Leroux-Roels et al. (2011): https://doi.org/10.1016/j.pervac.2011.05.005
Moussa et al. (2016): https://doi.org/10.1016/j.xphs.2015.11.002
Pratt (2018): https://doi.org/10.3390/antib7020019
Salazar et al. (2017): https://doi.org/10.1038/s41541-017-0019-3
Shankar et al. (2008): 10.1016/j.jpba.2008.09.020
Siegriest (2018): https://doi.org/10.1016/C2013-0-18914-3
Tovey & Lallemand (2011): https://doi.org/10.1177/2042098611406318
U.S National Library of Medicine: https://clinicaltrials.gov/ct2/home
Wang et al. (2014): https://doi.org/10.1016/j.bbrc.2014.07.090
Yasui et al. (2014): https://doi.org/10.1016/j.virol.2014.02.005
Images generated using Biorender.com
