Late-stage clinical trial attrition remains one of the most expensive challenges in drug development, with more than half of Phase III programs failing to meet their primary endpoints. In many cases, these failures are not driven by insufficient efficacy or safety, but by a more fundamental issue: the right biology was not measured in the right patients, at the right time. This is, at its core, a measurement problem.
Proteomics has long been positioned as a solution to this problem. Proteins are the functional effectors of disease biology, the direct targets of most therapeutics, and the most proximal molecular layer to clinical phenotype. Yet the translation of proteomic discovery into actionable clinical biomarkers remains inconsistent. In many cases, the limitation is not the technology itself, but how it is deployed. Different phases of drug development require fundamentally different measurement strategies, yet proteomic platforms are often used interchangeably across discovery, translation, and clinical testing.
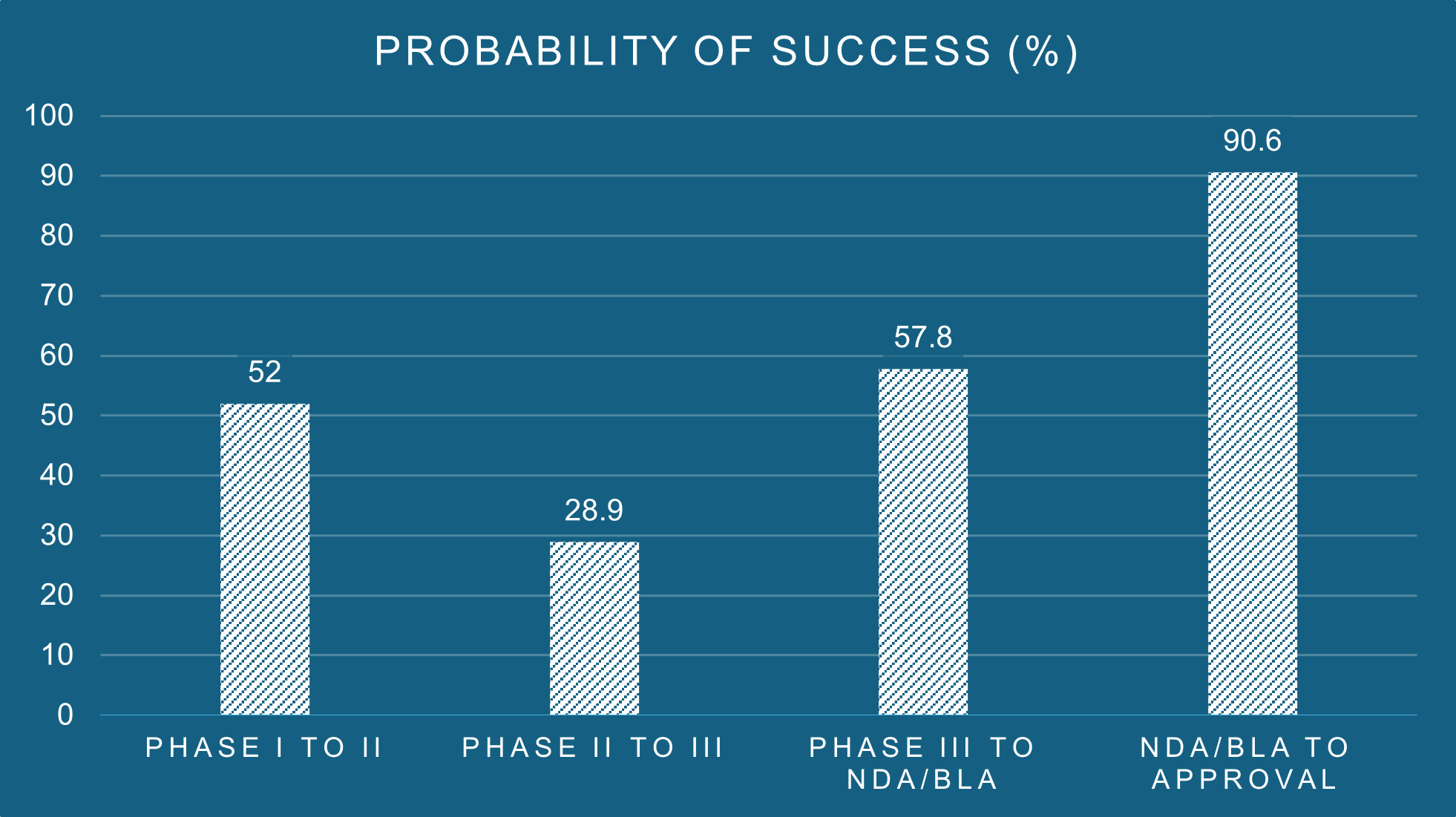
Figure 1: Overall phase transition success rates. Source: Biomedtracker® and Pharmapremia®, 2020.
The problem with premature commitment
Programs typically fail in one of two ways: they either commit too early to a narrow set of markers, or they never commit at all. The first involves nominating a small number of candidate proteins based on literature or preclinical data, and building a clinical strategy around them before patient heterogeneity is understood. The second is the inverse: relying on high-plex discovery platforms throughout development, generating large datasets without a defined analytical strategy or a clear path to clinical utility.
Both errors are costly. The first risks measuring the wrong proteins, missing the molecular signature that distinguishes responders from non-responders, or failing to detect an emergent safety signal in a protein not on the original panel. The second generates complexity without resolution, and rarely produces the analytically rigorous, reproducible measurements that clinical decision-making requires.
The solution is not a single platform. It is a principled, phase-appropriate integration of two complementary proteomic modalities.
Phase I: When breadth is the priority
Modern therapeutic modalities present a fundamental challenge for conventional biomarker strategies. Biologics targeting immune checkpoints, cytokine receptors, or co-stimulatory pathways are, by design, broadly immunomodulatory. Their effects are not confined to a single signalling axis but propagate across inflammatory, vascular, metabolic, and stromal compartments, varying substantially between patients, disease contexts, and stages of treatment. In this setting, the assumption that a small, predefined set of protein markers can capture the full biological response is rarely tested and often does not hold.
The limitation is not the underlying biology, but scale and heterogeneity. Published biomarker signals are typically derived from well-characterised, homogeneous cohorts, and do not always translate across the molecular diversity of Phase II or III populations. Patients with similar clinical diagnoses frequently represent distinct molecular endotypes, defined by the pathways driving disease rather than by symptoms alone. These endotypes respond differently to treatment, can exhibit different safety profiles, and remain largely invisible to targeted measurement strategies until sufficient breadth is applied to reveal them.
The second consequence of premature targeting is more subtle but equally important: the loss of covariance information. Proteins do not act in isolation. A circulating mediator elevated in one patient may reflect an acute inflammatory event; in another, chronic pathway dysregulation; in a third, a compensatory response to treatment. The same cytokine elevated in concert with a specific chemokine signature, a particular pattern of soluble receptor shedding, and the suppression of a counter-regulatory mediator tells an entirely different story, only visible when the network is measured. Targeted panels, by construction, remove that interpretive context before the investigator has established which context is relevant.
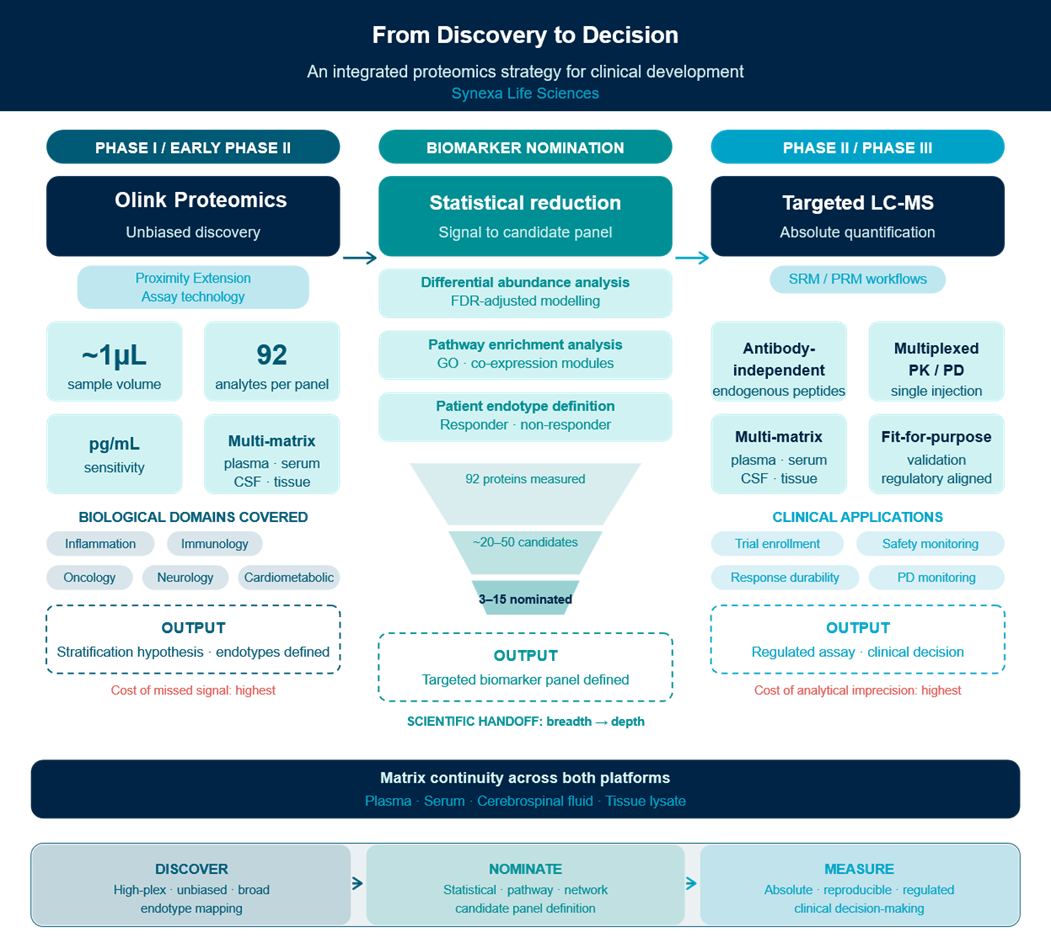
In early clinical development, the dominant requirement is coverage. This is precisely where a high-plex PEA-based proteomics platform like Olink is most informative. This approach delivers picogram-per-millilitre sensitivity across panels of up to 96 analytes per run, from as little as one microlitre of sample. Panels can be multiplexed across biological domains spanning inflammatory signalling, immune regulation, neurobiology, cardiovascular physiology, and oncology-relevant pathways, enabling simultaneous interrogation of the pathway landscape without a priori assumptions about which biology is operative in your population.
The output of this phase is not a clinical measurement. It is a nomination list: the subset of proteins that demonstrate statistically robust association with disease activity, treatment response, patient subgroup membership, or safety outcome. This is where stratification hypotheses are generated, molecular endotypes are defined, and the biological basis for trial enrichment is established.
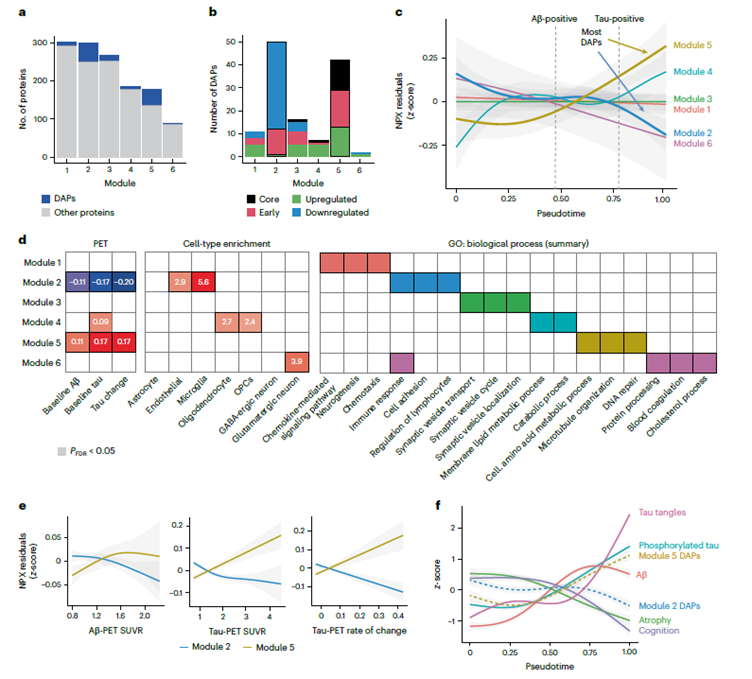
Figure 1: The practical value of broad proteomic coverage in defining disease endotypes is well illustrated by recent work in Alzheimer’s disease. Applying high-plex CSF proteomics across 877 deeply phenotyped participants spanning the full AD continuum, Binette and colleagues identified 127 differentially abundant proteins organised into distinct modules, each associated with different pathological stages, different cell types, and different biological processes. Critically, two opposing co-expression modules tracked the disease trajectory in divergent directions, a network-level finding that would have been entirely invisible to any targeted panel built from prior candidate biomarkers.
Adapted from Binette et al., 2024. Nature Neuroscience.
Phase II: From signal to measurement
Once candidate biomarkers are defined, the problem changes. The programme no longer needs breadth, it needs precision. The relevant proteins are known; what is now required is their accurate, absolute quantification at clinical throughput, across the longitudinal sample series that characterises a trial.
This transition is not merely operational. It reflects a genuine change in what the measurement must achieve. A cytokine signal identified in Phase I as part of a broader inflammatory signature may appear modest in isolation, but when quantified longitudinally in Phase II its trajectory can associate strongly with response durability, transforming a discovery-phase observation into a viable stratification marker. Equally, a protein linked to immune activation measured serially across a dosing cohort can reveal early divergence in patients who subsequently develop treatment-associated toxicity, enabling proactive safety monitoring before clinical events manifest. These require a different kind of measurement: absolute, reproducible, and anchored to a defined analytical standard.
This is precisely where many programmes introduce unnecessary complexity by treating their discovery platform as their validation platform. PEA technology is antibody-dependent by design, a deliberate architectural choice that confers the sensitivity and specificity required for broad discovery across diverse analyte classes. That same dependency, however, becomes a material consideration as programmes move toward regulated measurement. Antibody-based methods carry inherent lot-to-lot reagent variability, potential cross-reactivity in patients receiving biologic therapies, and a validation burden that increases substantially as assay use approaches regulatory decision-making.
Targeted liquid chromatography-mass spectrometry, configured for selected or parallel reaction monitoring, resolves these constraints at their source. By quantifying endogenous tryptic peptides directly, measuring the molecular entity itself rather than an antibody-dependent proxy, LC-MS provides absolute quantification that is reagent-independent, lot-invariant, and inherently specific to the analyte of interest. Multiplexed panels of three to fifteen nominated proteins can be measured in a single analytical run, with quantitative precision and inter-laboratory reproducibility that supports fit-for-purpose bioanalytical validation in alignment with established regulatory frameworks. The same targeted method applies across plasma, serum, cerebrospinal fluid, and tissue digest without platform transition, preserving the matrix continuity established during discovery and enabling programmes that evolve from peripheral blood sampling into mechanistic tissue or CNS work to carry their measurement strategy forward without interruption.
The result is a platform that does precisely what the clinical development stage demands: it measures a defined set of proteins, with absolute precision, at the throughput and reproducibility that clinical decision-making requires.
An integrated pipeline
The framing of high-plex immunoassay platforms and targeted LC-MS as competing technologies misunderstands both their design intent and their optimal deployment context. They are sequential tools in a coherent analytical pipeline, each performing the function it was built for. High-plex discovery identifies which proteins matter. Targeted LC-MS measures them with the precision and rigour that clinical development demands. The handoff between platforms is a scientifically principled decision point, where the breadth of discovery is deliberately contracted into the depth of measurement.
For biopharma teams building biomarker strategies from the ground up, the practical implication is clear. Invest in breadth early, when the cost of missing a relevant signal is highest. Transition to precision measurement once the signal is defined, when the cost of analytical imprecision is highest. And work with a partner capable of executing both phases with genuine scientific expertise, because the value of an integrated proteomics strategy is only fully realised when the discovery and measurement phases are designed as a single pipeline.
At Synexa, we use proteomic approaches to interrogate disease biology and treatment response in a structured, phase‑appropriate way, generating protein data that is biologically meaningful, analytically robust and aligned to stage of development. By combining proteomic data generation with scientific input on study design, biomarker selection and interpretation, we support the continuity of measurements as programmes progress through clinical development.
References
- BiomedTracker/PharmaPremia (Informa/BIO, 2020). Clinical Development Success Rates and Contributing Factors 2011–2020. Informa UK Ltd.
- Hay, M. et al. Clinical development success rates for investigational drugs. Nat. Biotechnol. 32, 40–51 (2014).
- Wong, C. H. et al. Estimation of clinical trial success rates and related parameters. Biostat. Oxf. Engl. 20, 273–286 (2019).
- Arrowsmith, J. & Miller, P. Phase II and phase III attrition rates 2011–2012. Nat. Rev. Drug Discov. 12, 569–569 (2013).
- Harrison, R. K. Phase II and phase III failures: 2013–2015. Nat. Rev. Drug Discov. 15, 817–818 (2016).
- Kim Y.C., Dean E., et al. Clinical development success rates for durable cell and gene therapies. Nature Reviews Drug Discovery. (2023).
- Binette et al. Proteomic changes in Alzheimer’s disease associated with progressive Aβ plaque and tau tangle pathologies. Nature Neuroscience. (2024).

Written by Caroline Beltran, Scientific Director
CBeltran@synexagroup.com
contactus@synexagroup.com
www.synexagroup.com